目的 回顾性分析脂质沉积性肌病(lipid storage myopathy,LSM)的临床及病理特点,提高临床中脂质沉积性肌病的诊断率,有助于治疗。方法 对15例临床上确诊的脂质沉积性肌病患者从临床、病理特点及治疗方面进行分析。结果 脂质沉积性肌病是脂肪酸氧化代谢障碍性疾病,临床主要表现为疲劳性肌无力,以近端肌肉为主(14例);激酶均有不同程度升高(15例);肌电图上呈肌源性损害(13例);抗线粒体抗体(AMA)免疫组化染色还可见到受损肌纤维内大量线粒体聚集;电镜下可见脂肪滴成串堆积在骨骼肌纤维中
脂质沉积性肌病的临床及病理分析
窦海玲1) 李世泽1)△ 吕海东2) 赵松耀1) 秦晓明1) 尹刘杰1)
1) 郑州大学附属郑州中心医院神经内科五病区,河南 郑州 450007 2)焦作市第一人民医院神经内科,河南 焦作 454002
作者简介:窦海玲,Email:douhailing2008@163.com
△通信作者:李世泽,Email:13683812815@163.com
【摘要】 目的 回顾性分析脂质沉积性肌病(lipid storage myopathy,LSM)的临床及病理特点,提高临床中脂质沉积性肌病的诊断率,有助于治疗。方法 对15例临床上确诊的脂质沉积性肌病患者从临床、病理特点及治疗方面进行分析。结果 脂质沉积性肌病是脂肪酸氧化代谢障碍性疾病,临床主要表现为疲劳性肌无力,以近端肌肉为主(14例);激酶均有不同程度升高(15例);肌电图上呈肌源性损害(13例);抗线粒体抗体(AMA)免疫组化染色还可见到受损肌纤维内大量线粒体聚集;电镜下可见脂肪滴成串堆积在骨骼肌纤维中。结论 脂质沉积性肌病在诊断上主要依靠临床、病理特点及生化检测;激素及维生素治疗可能有效。
【关键词】 脂质沉积性肌病;抗线粒体抗体;电镜;免疫组化;病理特点
【中图分类号】 R746 【文献标识码】 A 【文章编号】 1673-5110(2018)19-2031-05 DOI:10.12083/SYSJ.2018.19.464
Clinical and pathological analysis of lipid storage myopathy
DOU Hailing1),LI Shize1),LYU Haidong2),ZHAO Songyao1),QIN Xiaoming1),YIN Liujie1)
1) Department of Neurology,Zhengzhou Central Hospital Affiliated to Zhengzhou University,Zhengzhou 450007,China;2)Department of Neurology,First People's Hospital of Jiaozuo,Jiaozuo 454002,China
【Abstract】 Objective Retrospective analysis of the clinical and pathological features of lipid storage myopathy (LSM),improve the diagnostic rate of clinical deposition of lipid myopathy,and help treatment.Methods The clinical,pathological features and treatment of 15 clinically diagnosed patients with lipid deposition myopathy were analyzed.Results Lipid deposition myopathy is a fatty acid oxidative metabolic disorder disease,the main clinical manifestations of fatigue muscle weakness,mainly proximal muscle (14 cases);kinases are increased to varying degrees (15 cases);EMG Muscular damage (13 cases).Anti-mitochondrial antibody (AMA) immunohistochemical staining also showed a large number of mitochondria accumulation in the damaged muscle fibers;electron microscopically visible fat droplets accumulated in the skeletal muscle fibers.Conclusion Lipid deposition myopathy mainly relies on clinical,pathological features and biochemical tests in diagnosis;hormone and vitamin therapy may be effective.
【Key words】 LSM;AMA;Electronmicroscopy;Immunohistochemistry;Pathological Characteristics
脂质沉积性肌病(lipid storage myopathy,LSM)是肌肉能量代谢障碍性疾病中较常见的一种代谢性肌病。肌肉中脂肪代谢的任何一个环节出现问题都可导致肌肉脂肪代谢产能障碍,脂肪堆积在肌纤维间,肌肉组织的正常运作受到影响,机体产能不足出现一系列临床症状。临床上LSM主要表现为持续性肌无力(组织内脂质含量增加)和横纹肌溶解及运动不耐受(脂肪酸β氧化障碍)[1]。
近年来,随着诊疗条件的不断提高,尤其是分子生物学的广泛应用,人们对此类疾病有了更多的认识,分类越来越细,其目的是增加可治性肌病的检出率,提高患者生活质量,改善预后[2-3]。
1 资料与方法
1.1 对象与方法 患者选自郑州市中心医院与焦作市第一人民医院2000年至今经临床、病理及神经电生理确诊的LSM患者。按照一般情况、病程、首发症状、临床主要症状、肌酶、肌肉活检及治疗情况等几个方面进行整理分析。
1.2 临床特点
1.2.1 一般情况:15例LSM患者中男7例,女8例,发病年龄为12~57(25.6±15.75)岁,病程为1月~35 a。
1.2.2 首发症状:以发作性或波动性四肢无力为首发症状的11例;以双上肢无力起病的1例;以双下肢无力起病的2例;以恶心、呕吐等消化道症状起病的1例;其中2例首发症状同时伴其他症状,如肌肉压痛、纳差。
表1 15例LSM患者的临床资料
Table 1 Clinical data of 15 patients with LSM
| 编号 |
年龄/性别 |
病程 |
症状与体征 |
| 首发症状 |
吞咽困难 |
消化症状 |
循环系统 |
咀嚼困难 |
颈肌无力 |
| 1 |
35岁/女 |
12 a |
四肢无力 |
- |
+ |
- |
- |
- |
| 2 |
29岁/女 |
8 a |
四肢无力 |
+ |
- |
- |
- |
- |
| 3 |
20岁/女 |
3 a |
双下肢无力 |
- |
- |
+ |
- |
- |
| 4 |
47岁/男 |
5 a |
双上肢无力 |
- |
- |
- |
- |
- |
| 5 |
27岁/男 |
3月 |
双下肢无力 |
+ |
- |
- |
+ |
- |
| 6 |
23岁/男 |
6月 |
四肢无力 |
- |
+ |
- |
+ |
+ |
| 7 |
21岁/男 |
1月 |
四肢无力;肌肉压痛 |
+ |
- |
+ |
- |
+ |
| 8 |
40岁/男 |
5 a |
四肢无力;肌肉酸困 |
+ |
+ |
- |
+ |
+ |
| 9 |
35岁/男 |
8 a |
四肢无力 |
+ |
- |
- |
+ |
- |
| 10 |
17岁/男 |
5 a |
四肢无力 |
+ |
+ |
- |
+ |
+ |
| 11 |
57岁/男 |
35 a |
恶心、呕吐 |
- |
+ |
- |
- |
- |
| 12 |
30岁/女 |
4 a |
四肢无力 |
- |
- |
- |
- |
- |
| 13 |
46岁/女 |
13 a |
四肢无力 |
- |
- |
- |
- |
- |
| 14 |
36岁/女 |
5月 |
四肢无力 |
- |
- |
- |
- |
- |
| 15 |
12岁/女 |
1a |
四肢无力 |
- |
- |
- |
- |
- |
1.2.3 临床表现:15例患者均表现为骨骼肌无力,特点为近端无力为主,不耐疲劳。几乎均慢性起病,波动性进展,休息后可缓解。其中合并咀嚼无力5例,肌肉酸痛2例,合并消化道症状5例;心脏症状(心慌、心悸)2例,颈肌无力4例
1.3 辅助检查 血生化及神经电生理:15例LSM患者中均有不同程度的血清磷酸激酶升高及乳酸脱氢酶升高。15例LSM中13例为肌源性损害。1例为混合性损害。
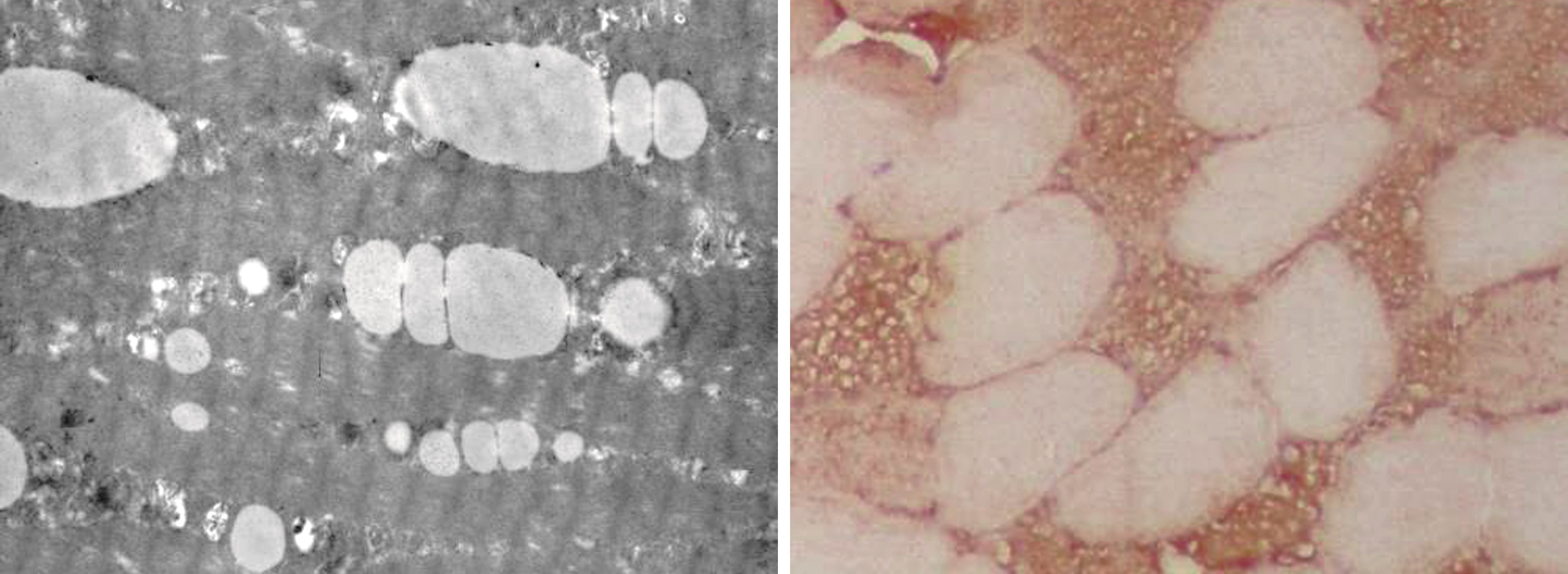
1.4 病理特点 对以上15例患者进行肌肉活检,几乎均表现为典型的LSM及肌肉病理改变。HE染色下可见染色肌纤维内空泡、筛孔样改变(图1A);油红“O”染色可见对应筛孔状肌纤维内大量脂质沉积性,被染成橘红色颗粒(图1B);抗线粒体抗体免疫组化染色下可见病变肌纤维内大量线粒体聚集(图2)。电镜下可见脂质沉积肌纤维膜下,呈串珠样(图2)。
图1 A:HE染色,肌纤维呈明显的空泡样改变 HE×400;B:油红“O”染色,筛孔状肌纤维内大量脂质沉积性,被染成橘红色颗粒×400
Figure 1 A:HE staining,Muscle fibers show obvious vacuolization changes HE× 400;B:Oil red“O” staining,a large amount of lipid deposition in the mesh-like muscle fibers,dyed into orange-red particles × 400
图2 A:电镜,增多的脂滴成串珠状排列于肌原纤维或肌浆膜下 EM×6 000;B:AMA免疫组化染色,肌纤维内大量线粒体聚集×400
Figure 2 A:Electron microscopy,increased lipid droplets are arranged in a beaded shape under the myofibrils or sarcolemma EM×6 000;B:AMA immunohistochemical staining,massive mitochondrial aggregation in muscle fibers× 400
2 治疗
5例确诊后自动出院,未治疗。3例确诊后给予激素静脉应用同时配合三磷酸腺苷、辅酶等药物治疗;并建议高碳水化合物、低脂肪饮食,症状好转。7例要求回去饮食控制。
3 讨论
脂质沉积性肌病是一组脂肪酸氧化代谢酶异常导致脂质蓄积于肌细胞而产生的一组临床病理综合征。LSM不是某一独立疾病,而是一组由于脂肪代谢障碍引起的一类疾病的统称。由于骨骼肌主要以脂肪酸为能量来源,当肌肉中脂肪代谢障碍时,便出现一系列症状。这也是肉碱存在于肌肉的主要原因[4-6]。
脂质沉积性肌病患者症状多为波动性,进行性进展,表现为运动不耐受,疲劳性肌无力,肌肉疼痛,这主要与肌糖原及葡萄糖大量消耗,而脂肪酸功能无法满足机体需要时出现疲劳性肌无力,休息后症状会有不同程度缓解[7-8]。受累肌群主要以近端骨骼肌和咀嚼肌为主。本组研究患者中,症状多局限于骨骼肌,均表现为发作性或波动性四肢无力,多下肢重于上肢,不耐疲劳,这可能与下肢负重大有关;部分患者还出现肌肉压痛、咀嚼肌受累,这可能与此类肌肉使用频率高有关。我们研究中有4例患者出现颈肌无力,有报道显示,若脊旁肌与咀嚼肌常常同时受累,临床上应高度怀疑LSM,此时应积极进行肌肉活检等明确诊断[9-12]。
脂质代谢贯穿人体整个机体,脂质代谢障碍除可累及肌肉系统外,也可累及全身各个系统。如神经、心脏、肝脏及胃黏膜等多个系统。这也可以解释患者常出现恶心、呕吐、心慌及心悸等症状[13-14]。33.3%患者出现消化道症状,这可能与胃肠道黏膜脂质沉积有关[15]。本组患者腱反射均降低,分析其机制可能与脂质沉积累及到雪旺细胞[16-17]。除以上临床症状外,LSM还可以以反复跌倒发作起病,极易误诊[18]。
LSM的病理上有其特点,HE染色上可见肌纤维间散在部分萎缩肌纤维,其间有大量筛孔样小泡,部分已融合成不规则的大空泡。油红“O”染色显示肌纤维内的空泡为大量脂肪颗粒充填。抗线粒体抗体免疫组化染色可见空泡内有大量线粒体聚集。分析可能与线粒体代偿性增生有关。因此,脂滴增多并发生融合可能成为脂质沉积性肌病的病理学特征[19-20]。目前有研究证实,受累肌肉中沉积的脂肪酸为甘油三酯。
虽然LSM病理特征基本相似,临床症状及治疗却有明显异质性,这与遗传背景及生化机制密切相关,目前LSM的命名趋于精准化[21-23]。电子显微镜下可见到排列成串珠样大小不一的脂滴沉积在受累肌纤维间,肌膜下及胞核周围。抗线粒体抗体免疫组化染色证实脂质周围出现线粒体增多[24]。
除以上临床及实验室特点外,影像磁共振检查对于LSM的早期筛查提供指导。LSM在磁共振上表现为脂质沉积改变的短T1长T2信号及肌纤维坏死改变的等T1长T2信号[25-27]。
由于该病累及范围广,症状缺乏特异性,临床症候常常多变,不易捕捉确诊,其中有5例就诊时诊断为多发性肌炎,3例初次就诊是误诊为慢性胃肠炎,2例就诊是误诊为心肌炎。此外,由于患者疲劳性肌无力,应积极与重症肌无力鉴别[28-29]。
有效及时的病理分析,将大大提高确诊率。由于激素可以影响病理特点,所以在病理检查前不应用激素。近期分子生物学的开展为本病的治疗提供了有力依据,对于LSM中某些亚型如核黄素反应性脂质沉积性肌病,通过分子基因分析,可以为后续治疗提供理论依据[30-32]。
本组患者有3例采用激素、配合能量制剂及饮食治疗,症状获得有效改善,这可能与激素可增加肾上腺素的脂质分解,促进脂肪酸线粒体内氧化有关[33-35]。
4 参考文献
[1] LAFORET P,VIANEY-SABAN C.Disorders of muscle lipid metabolism:diagnostic and therapeutic challenges[J].Neuromuscul Disord,2010,20(11):693-700.
[2] LIANG W C,NISHINO I.Lipid storage myopathy[J].Cur Neorol Neurosci Rep,2011,11(1):97-103.
[3] 张成,朱瑜龄.应重视可治性神经肌肉病的早期诊断与治疗[J].中国现代神经疾病杂志,2018,18(8):557-562.
[4] 野中征哉.临床肌肉病理学[M].吴士文,马维娅主译.3 版.北京:人民军医出版社,2007:104-109.
[5] 牛军伟.脂质沉积性肌病的临床、病理与基因学研究[D].中国人民解放军医学院,2015.
[6] AGUENNOUZ M,BECCARIA M,PURCARO G,et al.Analysis of lipid profile in lipid storage myopathy[J].J Chromatogr B Analyt Technol Biomed Life Sci,2016,1 029-1 030:157-168.doi:10.1016/j.jchromb.2016.06.039.
[7] ANGELINI C,TAVIAN D,MISSAGLIA S.Heterogeneous Phenotypes in Lipid Storage Myopathy Due to ETFDH Gene Mutations[J].JIMD Rep,2018,38:33-40.doi:10.1007/8904_2017_27.
[8] BERAADO A,DIMAURO S,HIRANO M.A diagnostic algorithm for metabolic myopathies[J].Curr Neurol Neurosci Rep,2010,10(2):118-126.
[9] 温冰.脂质沉积性肌病的临床、生化、分子病理和药物治疗研究[D].山东大学,2012.
[10] ANGELINI C,NASCIMEBNI A C,CENACCHI G,et al.Lipolysis and lipophagy in lipid storage myopathies[J].Biochim Biophys Acta,2016,1 862(7):1 367-1 373.
[11] 刘鹏,蒲传强.脂质沉积性肌病研究进展[J].中国误诊学杂志,2009,9(21):5 055-5 056.
[12] CHANDRA S R,CHRISTOPHER R,NARAYANAPPA G,et al.Lipid Storage Myopathy with Ketonuria:A Case of Fatty Acid Oxidation-Related Myopathy and Encephalopathy due to Multiple Acyl-CoA Dehydrogenase Deficiency[J].J Pediatr Neurosci,2018,13(3):362-365.doi:10.4103/JPN.JPN_21_18.
[13] FLANAGAN J L,SIMMONS P A,VEHIGE J,et al.Role of carnitine in disease[J].Nutr Metab,2010,16(7):30.
[14] STANLEY C A.New genetic defects in mitochondrial fatty acid oxidation and carnitine deficiency[J].Adv Pediatr,1987,34:59-88.
[15] VOCKLEY J,WHITEMAN D A.Defects of mitochondrial β oxidation:a growing group of Disorders[J].Neuromuscul Disord,2002,12(3):235-246.
[16] ANGELINI C,FREDDO L,BATTISTELLA P,et al.Carnitine palmityl transferase deficiency:Clinical variability,carrier detection,and autosomal-recessiv inheritance[J].Neurology,1981,31(7):883-886.
[17] 张燕,张博爱,瞿千千,等.脂质沉积性肌病的临床和神经电生理特征[J].郑州大学学报(医学版),2013,48(2):275-276.
[18] 马明明,王雪晶,丁雪冰,等.以反复跌倒发作为表现的脂质沉积性肌病临床及病理特征分析[J].中国实用神经疾病杂志,2014,17(23):19-21.
[19] 鄢传祝,王书运,刘淑萍,等.代谢性肌病的超微病理诊断[J].电子显微学报,1999,18(3):326-331.
[20] AGUENNOUZ M,BECCARIA M,PURCARO G,et al.Analysis of lipid profile in lipid storage myopathy[J].J Chromatogr B Analyt Technol Biomed Life Sci,2016,1:1 029-1 030.
[21] 吴志英,王柠.我国核黄素反应性脂质沉积性肌病基因研究的现状及热点问题[J].中华神经科杂志,2010,44(5):297-299.
[22] LIU X Y,JIN M,WANG Z Q,et al.Skeletal Muscle Magnetic Resonance Imaging of the Lower Limbs in Late-onset Lipid Storage Myopathy with Electron Transfer Flavoprotein Dehydrogenase Gene Mutations[J].Chin Med J (Engl),2016,129(12):1 425-1 431.doi:10.4103/0366-6999.183423.
[23] REILICH P,HORVATH R,KRAUSE S, et al.The phenotypic spectrum of neutral lipid storage myopathy due to mutations in the PNPLA2 gene[J].J Neurol,2011,258(11):1 987-1 997.doi:10.1007/s00415-011-6055-4.
[24] 周正平,肖 欣.脂质沉积性肌病的电镜超微病理诊断[J].电子显微学报,2016,35(3):258-260.
[25] ROSOW L K,AMATO A A.The Role of Electrodiagnostic Testing,Imaging,and Muscle Biopsy in the Investigation of Muscle Disease[J].Continuum(Minneap Minn),2016,22(6):1 787-1 802.
[26] GAETA M,MINUTOLI F,TOSCANO A,et al.Opposed-phase MR imaging of lipid storage myopathy in a case of Chanarin-Dorfman disease[J].Skeletal Radiol,2008,37(11):1 053-1 0537.doi:10.1007/s00256-008-0559-8.
[27] 郑贤应,慕容慎行,李银官,等.脂质沉积性肌病的MRI诊断[J].中国临床医学影像杂志,2006,17(8),458-460.
[28] ANGELINI C,NASCIMEBNI A C,CENACCHI G,et al.Lipolysis and lipophagy in lipid storage myopathies[J].Biochim Biophys Acta,2016,1 862(7):1 367-1 373.
[29] 程倚萌,周明锴,耿世平,等.炎性肌病脂质沉积性肌病肌电图结果对比分析[J].中国实用神经疾病杂志,2016,19(4):91-92.
[30] GOH L L,LEE Y,TAN E S,et al.Patient with mul-tiple acyl-CoA dehydrogenase deficiency disease and ETFDH mutations benefits from riboflavin therapy:a case report[J].BMC Med Genomics,2018,11(1):37.
[31] ANGELINI C,TAVIAN D,MISSAGLIA S.Heterogeneous Phenotypes in Lipid Storage Myopathy Due to ETFDH Gene Mutations[J].JIMD Rep,2018,38:33-40.doi:10.1007/8904_2017_27.Epub 2017 Apr 30.
[32] OHKUMA A,NONAKA I,MALICDAN M C,et al.Distal lipid storage myopathy due to PNPLA2 mutation[J].Neuromuscul Disord,2008,8(8):671-674.doi:10.1016/j.nmd.2008.06.382.
[33] BOSMA M.Lipid homeostasis in exercise[J].Drug Discov Today,2014,19(7):1 019-1 023.
[34] WANDERS R J,VREKEN P,DEN BOER M E,et al.Disorders of mitochondrial fatty acyl-CoA betaoxid-ation[J].J Inherit Metab Dis,1999,22(4):442-887.
[35] DE VISSER M,SCHOLTE H R,SCHUTGENS R B,et al.Riboflavin-responsive lipid-storage myopathy and glutaric aciduria type II of early adult onset[J].Neurology,1986,36(3):367-372.
(收稿2018-08-09 修回2018-09-29)
本文责编:关慧
本文引用信息:窦海玲,李世泽,吕海东,赵松耀,秦晓明,尹刘杰.脂质沉积性肌病的临床及病理分析[J].中国实用神经疾病杂志,2018,21(19):2131-2135.DOI:10.12083/SYSJ.2018.19.464
Reference information:DOU Hailing,LI Shize,LYU Haidong,ZHAO Songyao,QIN Xiuoming,YIN Liujie.Clinical and pathological analysis of lipid storage myopathy[J].Chinese Journal of Practical Nervous Diseases,2018,21(19):2131-2135.DOI:10.12083/SYSJ.2018.19.464