目的 探讨由ETFDH基因突变引起的核黄素反应性脂质沉积性肌病(Lipid storage myopathy,LSM)的临床特点以及核黄素的治疗效果。方法 通过肌肉磁共振(MRI)、肌肉活检及基因检测确诊两家系3例LSM患者,并评估给予核黄素治疗前后患者运动症状的变化。结果 2例左侧三角肌肌肉MRI与肌肉活检提示肌肉脂质沉积,基因检测揭示1例存在ETFDH基因c.1395dupT(p.G466Wfs*24)和c.770A>G(p.Y257C)复合杂合突变,2例存在ETFDH基因c.770A>G(p.Y257C)纯合突变,3例经核黄素治疗1个月后运动症状均得到显著改善。结论 对于无明显诱因引起的双下肢无力患者,可通过肌肉活检和基因检测进行确诊,且核黄素能够有效改善运动症状。
核黄素反应性脂质沉积性肌病两家系临床分析
陈永康 刘 晗 袁 心 王雪晶 滕军放△
郑州大学第一附属医院神经内科,河南郑州 450052
基金项目:国家自然科学基金(编号:81671267)
作者简介:陈永康,Email:magic0328520@163.com
△通信作者:滕军放,Email:13838210077@163.com
【摘要】 目的 探讨由ETFDH基因突变引起的核黄素反应性脂质沉积性肌病(Lipid storage myopathy,LSM)的临床特点以及核黄素的治疗效果。方法 通过肌肉磁共振(MRI)、肌肉活检及基因检测确诊两家系3例LSM患者,并评估给予核黄素治疗前后患者运动症状的变化。结果 2例左侧三角肌肌肉MRI与肌肉活检提示肌肉脂质沉积,基因检测揭示1例存在ETFDH基因c.1395dupT(p.G466Wfs*24)和c.770A>G(p.Y257C)复合杂合突变,2例存在ETFDH基因c.770A>G(p.Y257C)纯合突变,3例经核黄素治疗1个月后运动症状均得到显著改善。结论 对于无明显诱因引起的双下肢无力患者,可通过肌肉活检和基因检测进行确诊,且核黄素能够有效改善运动症状。
【关键词】 脂质沉积性肌病;ETFDH基因;肌肉活检;核黄素;基因检测
【中图分类号】 R596 【文献标识码】 A 【文章编号】 1673-5110(2018)24-2692-05 DOI:10.12083/SYSJ.2018.24.562
Clinical analysis of riboflavin-reactive lipid storage myopathy in two families
CHEN Yongkang,LIU Han,YUAN Xin,WANG Xuejing,TENG Junfang
Department of Neurology,the First Affiliated Hospital of Zhengzhou University,Zhengzhou 450052,China
【Abstract】 Objective To investigate the clinical features of riboflavin-reactive lipid storage myopathy (LSM) caused by ETFDH gene mutation and the therapeutic effect of using riboflavin.Methods Three patients in two family with LSM were diagnosed by muscle magnetic resonance imaging (MRI),biopsy,and genetic testing,and the changes in motor symptoms before and after treatment with riboflavin were evaluated.Results In both 3 patients,MRI and muscle biopsy of the left deltoid muscle showed muscle lipid deposition.The gene detection revealed the presence of the complex heterozygous mutation c.1395dupT (p.G466Wfs*24) and c.770A>G(p.Y257C) in the ETFDH gene in one patient,and two patients carry the c.770A>G(p.Y257C) homozygosis mutation in the ETFDH gene,and significant improvement in motor symptoms were observed after 1 month of treatment with riboflavin in 3 patients.Conclusion This report suggests that patients with dual lower limb weakness caused by no obvious cause can be diagnosed by muscle biopsy and genetic testing,and the application of riboflavin can effectively improve the patient's motor symptoms.
【Key words】 Lipid storage myopathy;ETFDH gene;Muscle biopsy;Riboflavin;Genetic Testing
脂质沉积性肌病(lipid storage myopathy,LSM)是一种常染色体隐性遗传病,以肌肉中异常的脂质沉积为其病理特征。虽然进行了广泛的分子研究,但只有4种类型可遗传诊断为LSM,包括原发性肉碱缺乏症(PCD)、多种脂酰辅酶A缺乏症(MADD)、中性脂肪贮存伴鱼鳞病(NLSDI)和中性脂肪贮存性肌病(NLSDM)[1]。本次研究报道两家系3例经肌肉核磁共振(MRI)、肌肉活检及基因检测确诊的核黄素反应性LSM患者,并结合相关文献分析其临床特点及使用核黄素的治疗效果。
1 资料与方法
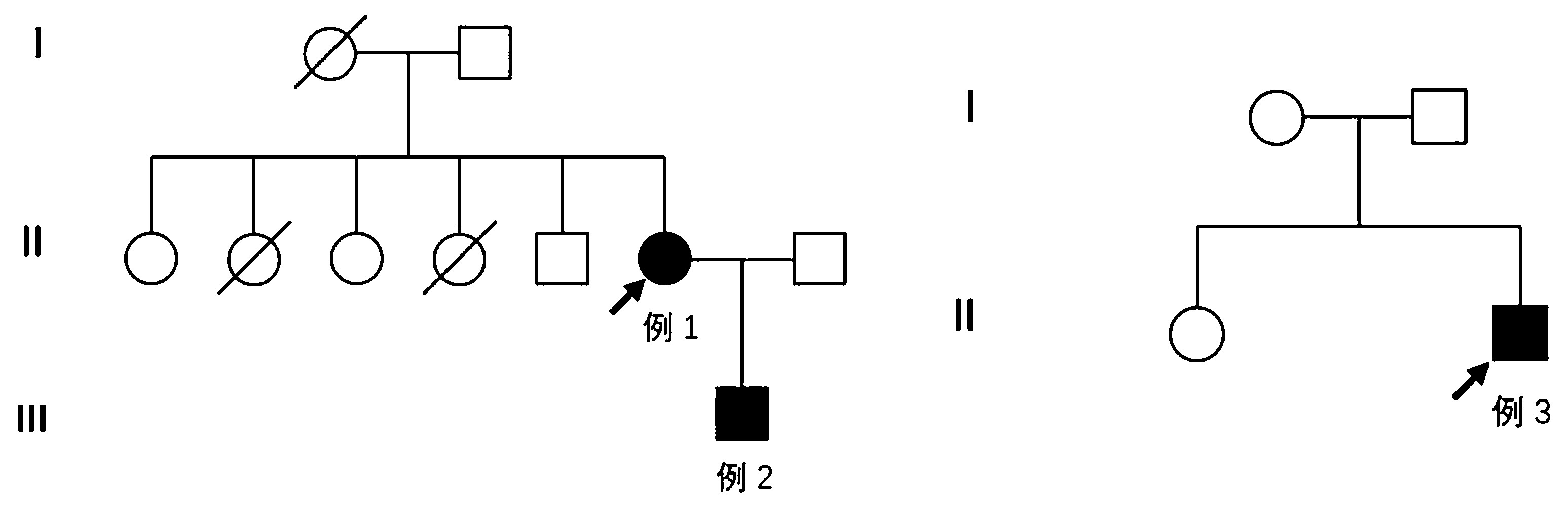
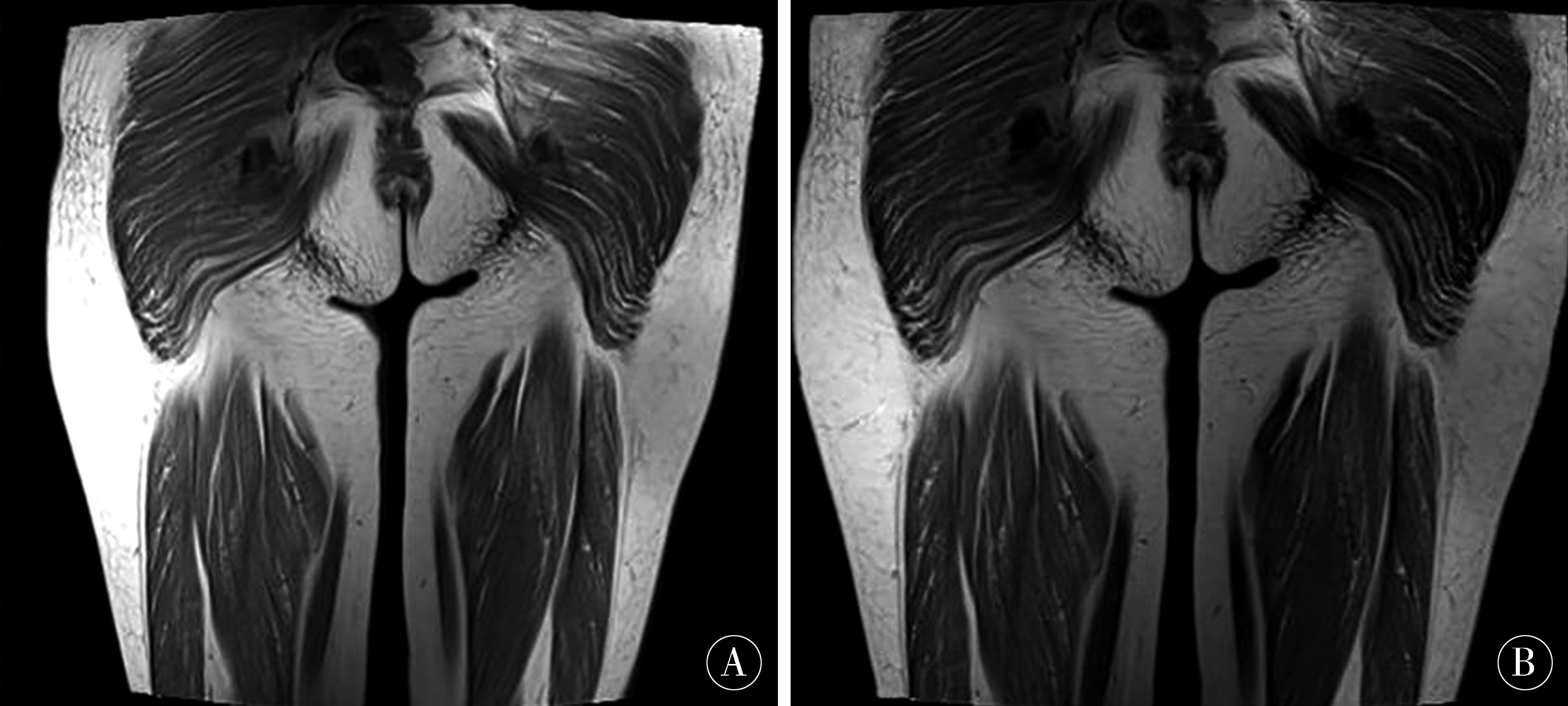
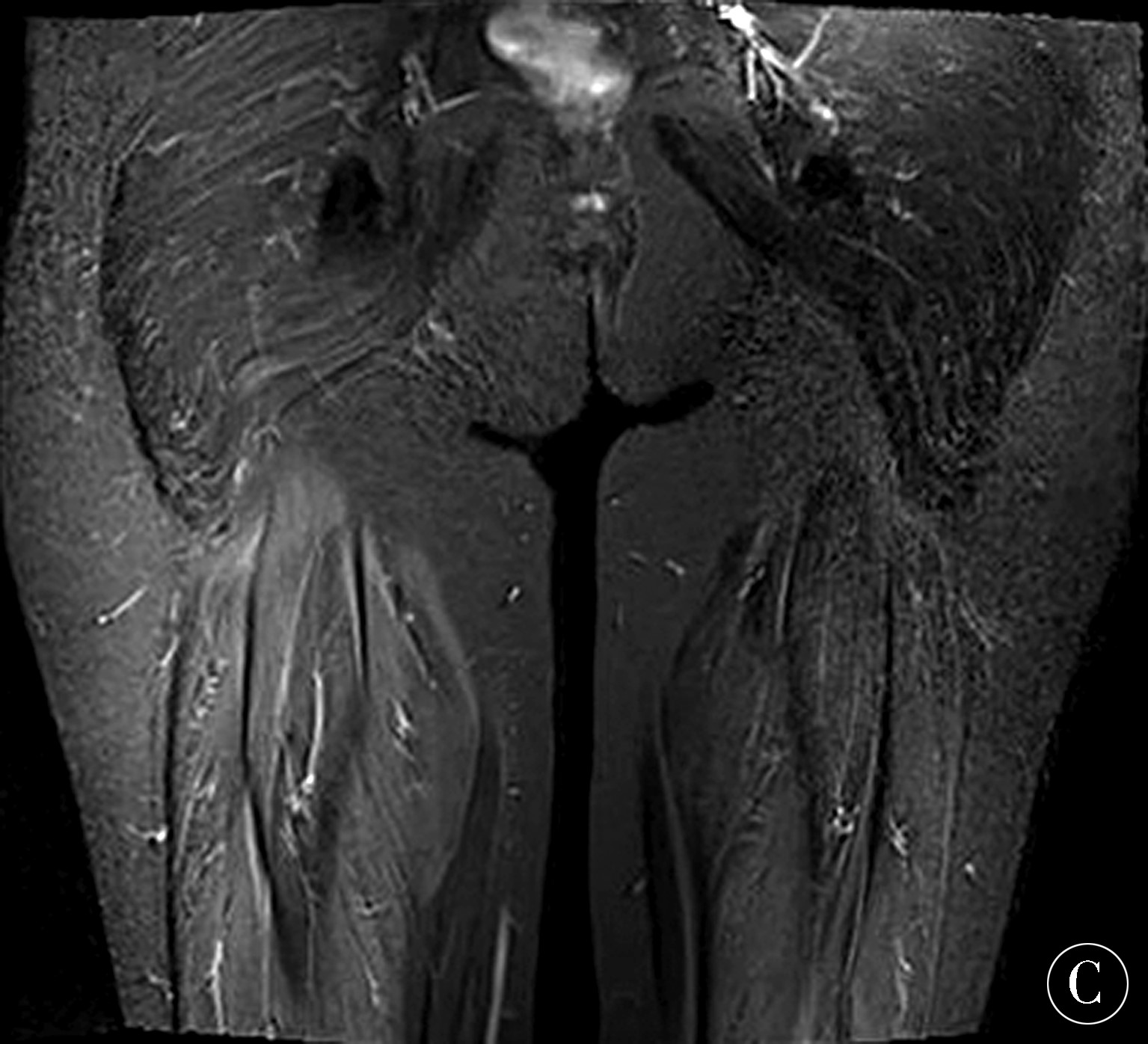
1.1 一般资料 患者家系图如图1所示。病例1女,48岁,因“行走无力10年余”于2018-04-03就诊于郑州大学第一附属医院。10a前患者无明显诱因出现四肢无力、行走困难、头部抬举困难症状,自诉口服肌苷片、丹参药物后症状逐渐缓解。3个月前无明显诱因出现双下肢无力,行走困难,感头重脚轻,心前区不适,伴双上肢逐渐出现无力症状,持物不稳,抬举稍困难、抬举后呈强制性仰位,余无特殊不适。遂至当地医院,查心肌酶谱示:肌红蛋白162.2 ng/mL(↑),肌酸激酶同工酶13.46 ng/mL,肌酸激酶361 ng/mL(↑),乳酸脱氢酶600U/L(↑),体格检查示神志清楚,精神可,脑神经检查未见明显异常,双上肢肌力5级,双下肢肌力4级,右侧轻瘫试验阳性,四肢腱反射正常(++),双侧病理征阳性。行胸片、心电图、心脏彩超、冠脉CTA未见明显异常,双下肢近端肌肉MRI检查(图2)示:双侧臀部及双侧大腿后部肌群内可见短T、1长T2信号,其中右侧大腿后部肌群呈压脂稍高信号,提示:右侧大腿后部肌群轻度水肿,双侧臀部及双侧大腿后部肌群内脂肪沉积。
图1 患者家系图
Figure 1 Patient family diagram
图2 病例1下肢近端肌肉MRI A:双侧臀部及双侧大腿后部肌群短T1像;B:双侧臀部及双侧大腿后部肌群长T2像;C:双侧臀部及双侧大腿后部肌群压脂像,其中右侧大腿肌群压脂信号稍高
Figure 2 Case 1 MRI of the proximal muscles of the lower extremities A:short T1 images of the bilateral buttocks and the muscles of the posterior thighs of both sides;B:T2 images of the muscles of the posterior muscles of both sides of the hips and the sides of the thighs;C:bilateral buttocks and bilateral thighs The posterior muscle group is fat-pressed,in which the right thigh muscle group has a slightly higher pressure signal
病例2男,20岁,因“运动后疲劳感2年余,加重1个月”于2018-04-03就诊于郑州大学第一附属医院。2 a前患者无明显诱因情况下出现行走时双下肢无力,运动后明显疲劳感。就诊于当地医院,给予激素治疗后可缓解,但不及正常人,1个月前因受凉后病情加重,出现四肢近端肢体无力。实验室检查心肌酶谱示:肌红蛋白143.4 ng/mL(↑),肌酸激酶同工酶43.79 ng/mL(↑),肌酸激酶451 ng/mL(↑),乳酸脱氢酶823 U/L(↑),体格检查示生命体征平稳,神志清楚,脑神经检查未见明显异常,四肢近端肌力4级,四肢腱反射正常(++),病理征阴性。行胸片、心电图、心脏彩超等检查未见明显异常。
病例3男,16岁,因“四肢无力,伴体质量下降1 a”于2018-05-23就诊于郑州大学第一附属医院。1 a前患者无明显诱因出现渐进性四肢无力,近1 a体质量减轻5.0 kg。发病以来,未给予治疗。实验室检查心肌酶谱示:肌红蛋白137.9 ng/mL(↑),肌酸激酶同工酶48.65 ng/mL(↑),肌酸激酶582 ng/mL(↑),乳酸脱氢酶945 U/L(↑),体格检查示生命体征平稳,神志清楚,精神差,脑神经检查未见明显异常,四肢近端肌力4级,四肢肌轻微萎缩,四肢腱反射稍低,病理征阴性。行胸片、心电图、心脏彩超等检查未见明显异常。
1.2 肌肉病理检查方法 局麻下取3例左侧三角肌进行伊红苏木素(HE)染色,改良Gomori(MGT)染色,油红 “ O”(ORO)染色及三磷酸腺苷酶 (ATP)染色,于光镜下观察肌肉组织病理变化。
1.3 基因检测 采集3例患者的外周血样进行Sanger测序分析发现,1例存在ETFDH基因c.1395dupT(p.G466Wfs*24)和c.770A>G(p.Y257C)复合杂合突变,2例存在ETFDH基因c.770A>G(p.Y257C)纯合突变。对其余家庭成员的血样进行Sanger测序分析,发现病例1父亲携带有ETFDH基因c.1395dupT(p.G466Wfs*24)杂合突变,病例1丈夫携带有ETFDH基因c.770A>G(p.Y257C)杂合突变,病例3父亲和母亲均携带有ETFDH基因c.770A>G(p.Y257C)杂合突变。
2 结果
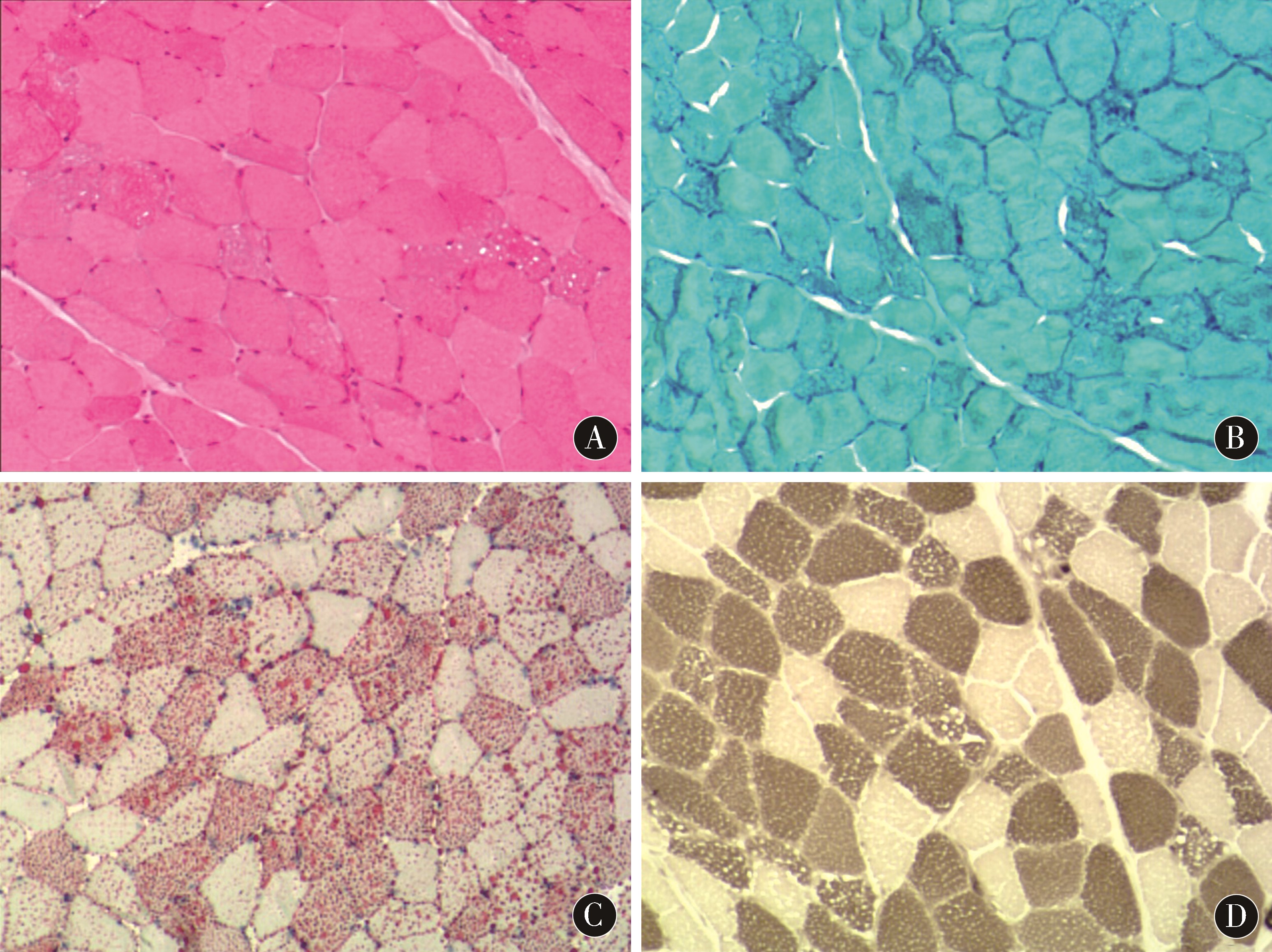
2.1 肌肉病理学特点 病例1左侧三角肌肌肉病理 (图3)示:HE染色显示肌纤维轻度大小不一,少量肌纤维萎缩,部分肌纤维可见多量小空泡;MGT染色显示部分纤维胞浆内可见小空泡;ORO染色显示胞浆内空泡呈红色;ATP染色显示Ⅰ型Ⅱ型肌纤维大致相等,两者镶嵌分布。病例2和病例3左侧三角肌肌肉具有相似的病理结果。
图3 病例1左侧三角肌肌肉活检 A:HE染色(×200)显示肌纤维轻度大小不一,少量肌纤维萎缩部分肌纤维可见多量小空泡;B:MGT染色(×200)显示部分纤维胞浆内可见小空泡;C:ORO染色(×200)显示胞浆内空泡呈红色;D:ATP染色(×200)显示Ⅰ型、Ⅱ型肌纤维大致相等,两者镶嵌分布
Figure 3 Case 1 left deltoid muscle biopsy A:HE staining (×200) showed that the muscle fibers were slightly different in size,a small amount of muscle fibers were atrophied,and some muscle fibers were visible in small amounts of small vacuoles;B:MGT staining (×200) showed small vacuoles in some fiber cytoplasm bubble;C:ORO staining (×200) showed red vacuoles in the cytoplasm;D:ATP staining (×200) showed that type Ⅰ muscle fibers were approximately equal to type Ⅱ,with mosaics
2.2 基因突变 分析通过Sanger测序发现携带ETFDH基因c.770A>G(p.Y257C)的纯合突变和复合杂合突变的家庭成员具有LSM的相关表型,而仅携带ETFDH基因c.770A>G(p.Y257C)杂合突变或c.1395dupT(p.G466Wfs*24)杂合突变的家庭成员未表现出任何LSM相关临床症状。在中国人群中已有ETFDH基因复合杂合突变和纯合突变致病的文献报道[2]。c.770A>G(p.Y257C)变异为错义突变,在正常人群中携带频率极低,已有文献报道该位点变异与LSM相关[3],与我们报道中的患者情况相符;c.1395dupT(p.G466Wfs*24)为移码突变,可导致蛋白编码提前终止,为有害突变的可能性大,该位点在正常人群中携带频率极低,其致病性未见报道,临床意义未明。推测病例1母亲携带有EFFDH基因c.770A>G(p.Y257C)杂合突变,该家系患者LSM的临床表现主要是由EFFDH基因的c.770A>G(p.Y257C)错义突变导致的,而c.1395dupT(p.G466Wfs*24)移码突变是否为LSM的致病性突变还有待进一步的研究。
2.3 治疗结果 3例患者诊断为“脂质沉积性肌病”,给予核黄素磷酸钠等神经营养剂治疗,嘱患者高碳水化合物,低脂肪、低蛋白饮食,一日多餐,适当锻炼。2周后3例患者病情迅速好转,运动症状得到显著改善,1个月后,病例1下肢无力症状基本消失,病例2下肢肌力基本恢复正常,可从事正常的体力活动,疲劳感基本消失,病例3四肢肌力有所改善,体质量有所增加,但还未达到正常标准。
3 讨论
脂质被定义为一种疏水性生物分子,主要由两种类型的分子组成:脂肪(即甘油三酯)和类脂(固醇及其脂、磷脂和糖脂等)。脂质主要以三酰甘油的形式储存在肌纤维中,在正常情况下约占纤维体积的0.2%[4],其水解生成的脂肪酸最终通过线粒体基质中的β-氧化循环分解代谢,产生ATP为机体供能。催化脂肪酸氧化的酶系存在于线粒体基质,长链脂酰CoA不能直接透过线粒体内膜,需要借助肉碱脂酰转移酶Ⅰ,转化为脂酰肉碱进入线粒体。进入线粒体的脂酰肉碱在肉碱脂酰转移酶Ⅱ的作用下,转化为的脂酰CoA经过脱氢、加水、再脱氢、硫解,完成一次β-氧化循环。生成的FADH2、NADH经氧化呼吸链氧化,与ADP磷酸化偶联,产生ATP为机体功能。由ETFDH基因编码的黄素蛋白ETF-QO蛋白嵌合于线粒体的内膜上,它接受氧化呼吸链上游传递来的电子,通过下游辅酶Q10(CoQ10)的还原反应,将脂肪酸的氧化作用与线粒体呼吸作用联系在一起。
LSM是以脂质代谢异常引起的肌纤维中脂质积聚的疾病,生化发病机制常见于以下情况[5]:(1)当肉碱脂酰转移酶缺乏或者遭到破坏时,导致长链脂酰CoA不能进入线粒体,从而大量聚集于细胞中;(2)脂肪酸β-氧化脱氢阶段过程中主要参与的酶有酰基辅酶A脱氢酶家族,是一系列以黄素腺嘌呤二核苷酸(FAD)为辅基的黄素蛋白酶,所以当脂质代谢过程中的酶缺乏时会影响肌纤维内的脂质代谢,最终导致异常增多的脂肪在肌纤维内堆积而引起肌病;(3)由ETFDH基因编码的黄素蛋白ETF-QO蛋白功能障碍时,可导致氧化呼吸链中上游代谢产物聚集,降低脂肪酸的β-氧化,使各种链长度的脂酰肉碱在血中积聚,肌纤维出现大量无法代谢的脂肪滴。
脂质代谢通路中相关的酶或蛋白质的缺陷,导致脂肪酸代谢紊乱和细胞内脂肪沉积和细胞损伤,从而引起的复杂多样的临床表现,其中以肌肉受累最常见。脂肪代谢性肌病是一种渐进性疾病[3],主要有两种表现形式,一种表现为进行性肌无力和运动不耐受,肌肉活检可见大量的脂滴沉积,即LSM;另一种表现为在运动、感染、寒冷、饥饿或某些药物(大剂量安定、丙戊酸盐等)等应激条件下,诱发反复发作性横纹肌溶解伴肌红蛋白尿,而肌肉活检没有或极少有脂肪沉积。
本报道中的患者为第一种临床表现形式,以近端肌和躯干肌无力为主,病程呈波动性,发病时患者的双下肢近端肌肉MRI结果提示临床医生在缺乏肌肉活检和基因检测结果时,可以考虑用肌肉MRI成像作为诊断依据。肌肉MRI是神经肌肉疾病的辅助诊断工具,具有无创、方便的特点,下肢T1和T2加权轴向图像可用来诊断肌营养不良。已有文献证实LSM患者的大腿前、后、内侧肌群可表现出典型的脂肪浸润和萎缩,其中大腿后肌群的脂肪浸润评分明显高于大腿前肌群和大腿内侧肌群[6]。
有文献表明大多数中国LSM患者的临床症状是由ETFDH基因突变所致轻度的MADD引起的[7]。MADD患者线粒体中,由于电子转移黄素蛋白(ETF)或ETFDH基因缺陷,使黄素蛋白ETF-QO功能障碍,从而导致脂肪酸的β-氧化脱氢反应受损[3]。其临床类型可分为新生儿型和迟发型。新生儿型发病形式以低张力、肝肿大、非酮症低血糖和代谢性酸中毒为主,婴儿多于早期死亡[8]。迟发型的发病形式表现为,在一定诱因条件下出现以近端肌群为主的肌无力,肌肉病理示脂质沉积,血清肌酸激酶常明显或轻度增高。
通过特定的检查,做出准确的诊断对于LSM的治疗至关重要,越来越多的证据表明:服用高剂量的肉碱可以明显改善PCD患者的病情[9]。而对于ETFDH突变引起的MADD,核黄素的治疗是有效的,因为核黄素为FAD的前体物质,当补充核黄素时可增加线粒体内FAD浓度,从而促进黄素蛋白与FAD结合,改善突变引起的蛋白与FAD的亲和力。本报道中的3例患者经过核黄素治疗后,运动症状得到显著改善,这就提示我们在临床上,核黄素可用来治疗LSM,并且具有很好的效果。
核黄素反应性LSM在临床上是一种可通过药物治疗的遗传性疾病,ETFDH是LSM主要的致病基因。对于那些无明显诱因出现下肢近端肢体无力,运动不耐受的患者,可以通过肌肉MRI,肌肉活检以及基因检测手段进行确诊,并且可以通过给予核黄素进行有效治疗。
4 参考文献
[1] REILICH P,HORVATH R,KRAUSE S,et al.The phenotypic spectrum of neutral lipidstorage myopathy due to mutations in the PNPLA2 gene[J].J Neurol,2011,258(11):1987-1997.
[2] 操基清,张成,李亚勤,等.核黄素反应性脂质沉积性肌病临床特征与基因突变分析:两家系三例报告并文献复习[J].中国现代神经疾病杂志,2014,14(6):479-484.
[3] OLSEN R K,OLPIN S E,ANDRESEN B S,et al.ETFDH mutations as a major cause of riboflavin-responsive multiple acyl-CoA dehydrogenation deficiency[J].Brain,2007,130(Pt 8):2 045-2 054.
[4] ZHANG J,FRERMAN F E,KIM J J.Structure of electron transfer flavoprotein-ubiquinone oxidoreductase and electron transfer to the mitochondrial ubiquinone pool[J].Proc Natl Acad Sci U S A,2006,103(44):16 212-16 217.
[5] HUI A S,BAUER A L,STRIET J B,et al.Calcium signaling stimulates translation of HIF-alpha during hypoxia[J].FASEB J,2006,20(3):466-475.
[6] LIU X Y,JIN M,WANG Z Q,et al.Skeletal Muscle Magnetic Resonance Imaging of the Lower Limbs in Late-onset Lipid Storage Myopathy with Electron Transfer Flavoprotein Dehydrogenase Gene Mutations[J].Chin Med J (Engl),2016,129(12):1 425-1 431.
[7] WEN B,DAI T,LI W,et al.Riboflavin-responsive lipid-storage myopathy caused by ETFDH gene mutations[J].J Neurol Neurosurg Psychiatry,2010,81(2):231-236.
[8] OLSEN R K,ANDRESEN B S,CHRISTENSEN E,et al.Clear relationship between ETF/ETFDH genotype and phenotype in patients with multiple acyl-CoA dehydrogenation deficiency[J].Hum Mutat,2003,22(1):12-23.
[9] LIANG W C,NISHINO I.Lipid storage myopathy[J].Curr Neurol Neurosci Rep,2011,11(1):97-103.
(收稿 2018-09-12 修回2018-10-05)
本文责编:夏保军
本文引用信息:陈永康,刘晗,袁心,王雪晶,滕军放.核黄素反应性脂质沉积性肌病两家系临床分析[J].中国实用神经疾病杂志,2018,21(24):2692-2696.DOI:10.12083/SYSJ.2018.24.562
Reference information:CHEN Yongkang,LIU Han,YUAN Xin,WANG Xuejing,TENG Junfang.Clinical analysis of riboflavin-reactive lipid storage myopathy in two families[J].Chinese Journal of Practical Nervous Diseases,2018,21(24):2692-2696.DOI:10.12083/SYSJ.2018.24.562